Getting started

Help needed for gmx_MMPBSA! To the content creators out there...¶
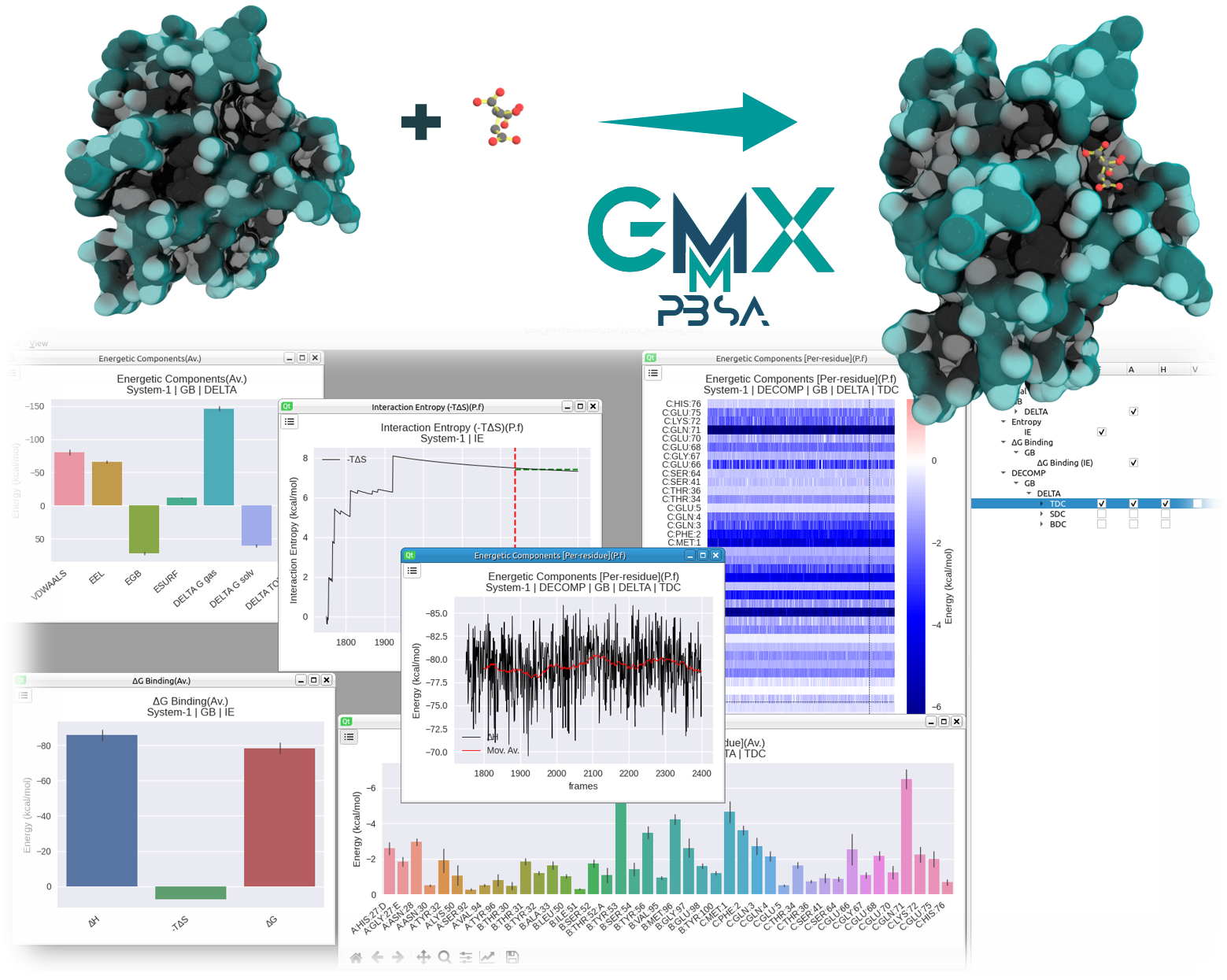
gmx_MMPBSA is a new tool based on AMBER's MMPBSA.py, aiming to perform end-state free energy calculations with GROMACS files. It works with all GROMACS versions along with AmberTools >=20 and brings improvements in compatibility, versatility, analyses, and parallelization compared to existing programs (see here for a detailed comparison)
Cite gmx_MMPBSA
gmx_MMPBSA official paper has been published on Journal of Chemical Theory and Computation and can be accessed here. If you use gmx_MMPBSA, please cite it as follows:
Valdés-Tresanco, M.S., Valdés-Tresanco, M.E., Valiente, P.A., and Moreno, E. gmx_MMPBSA: A New Tool to Perform End-State Free Energy Calculations with GROMACS. Journal of Chemical Theory and Computation, 2021 17 (10), 6281-6291. https://pubs.acs.org/doi/10.1021/acs.jctc.1c00645.
Please, also consider citing MMPBSA.py's paper:
Bill R. Miller, T. Dwight McGee, Jason M. Swails, Nadine Homeyer, Holger Gohlke, and Adrian E. Roitberg. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. Journal of Chemical Theory and Computation, 2012 8 (9), 3314-3321. https://pubs.acs.org/doi/10.1021/ct300418h.
Download | *.bib | *.ris | *.xml
Please, visit Cite gmx_MMPBSA page for more information on how to cite gmx_MMPBSA and the programs/methods implemented in it.
Installation¶
Ready to use gmx_MMPBSA 😀? Check the installation page
What can be done with gmx_MMPBSA?¶
Multiple calculations can be performed with gmx_MMPBSA, such as:
- Binding free energy calculations with PB, GB and/or 3D-RISM models
- Alanine scanning
- Binding free energy decomposition
- Entropy corrections (IE, C2, NMODE)
- Stability
- QM/MMGBSA
There is always more...
You can check gmx_MMPBSA in a nutshell page for a more detailed overview of the types of calculations supported in gmx_MMPBSA. Also, check our example page to see a detailed list of all the examples available
In the current version, gmx_MMPBSA supports a number of different systems, including but not limited to:
- Protein-protein
- Protein-ligand
- Protein-DNA
- Metalloprotein-ligand
- Protein-glycan
- Membrane proteins
- Multicomponent systems (e.g., Protein-DNA-RNA-Ions-Ligand)
Support for Amber, OPLS, and CHARMM force fields
In the current version, gmx_MMPBSA supports Amber, OPLS, and CHARMM force fields. Any system built for GROMACS with either pdb2gmx or CHARMM-GUI is supported in gmx_MMPBSA. Likewise, any system built for NAMD and potentially any other software that uses *.psf - *.dcd files can be processed in gmx_MMPBSA 😀. Check our example page to see a detailed list of all the examples available.
The following video shows how a typical binding free energy calculation with the GB model and the Interaction entropy method is performed in gmx_MMPBSA.
gmx_MMPBSA a quick overview¶
gmx_MMPBSA is a Python module that contains three applications:
- gmx_MMPBSA is the fundamental application and carries out the calculations mentioned above
- gmx_MMPBSA_ana provides an intuitive way to analyze the data from gmx_MMPBSA calculations and save high-quality pictures
- gmx_MMPBSA_test is a tool designed to test if the installation was successful by running one or more available examples in gmx_MMPBSA.
Easy to run
gmx_MMPBSA can run in parallel and requires only a few things to perform any calculation. That is:
- an input parameters file (
*.in, contains all the specifications regarding the type of calculation that is going to be performed) - a MD Structure+mass(db) file (
*.tpr,*.pdb) - an index file (
*.ndx) - receptor and ligand groups (group numbers in the index file)
- a trajectory file (
*.xtc,*.pdb,*.trr) - On certain occasions, defining a topology file (
*.top) may be required.
Once the calculation is done, you can analyze the results in gmx_MMPBSA_ana
You can check How gmx_MMPBSA works page to get more details. Also check our example page to see how gmx_MMPBSA works with real examples
If you prefer a more general overview of the gmx_MMPBSA suite, check this amazing video by Dr. Hymavathi Veeravarapu.
Need help?¶
Help section contains the most frequently asked questions and errors. Also, look at our Google group or the issues section to find out about specific cases and others.
If you still have doubts or cannot solve the problem, please consider opening an issue or posting in our Google group
Follow gmx_MMPBSA¶
Visit Pypi Stats or PePy to see how gmx_MMPBSA is doing.
Collaboration¶
gmx_MMPBSA is rapidly becoming one of the main programs to perform end-point free energy calculations out there (~29k downloads so far). We are currently focused on optimizing the program, supporting new types of calculations, force fields, etc. However, the video tutorials are not that great.
That being said, and here is where we need help, we will appreciate any collaboration in making video tutorials for the documentation. The video tutorial will be included as a link to YouTube in the gmx_MMPBSA documentation with full acknowledgment of the person who created the video tutorial. It will also be acknowledged as a contributor of gmx_MMPBSA... 😃
Feel free to contact us through our Google group (https://groups.google.com/g/gmx_mmpbsa) for more details...
Acknowledgments¶
- First of all, to Amber and GROMACS developers. Without their incredible and hard work, gmx_MMPBSA would not exist.
- Jason Swails (Amber developer and ParmEd principal developer) for his continuous support on ParmEd issues.
- Dr. Hymavathi Veeravarapu for helping with the introductory video for gmx_MMPBSA

- To the Open Source license of the JetBrains programs.
![]()
- To the Sourcery team for supporting us with the Pro version.
- To all researchers who help improve gmx_MMPBSA with comments, feedback, and bug reports.
Created: February 8, 2021 07:10:13