Running gmx_MMPBSA
Before running gmx_MMPBSA¶
gmx_MMPBSA requires minimum processing on the input structure and trajectory files. Before running gmx_MMPBSA, please make sure:
The structure defined in -cs, -rs, or -ls options is consistent
Visualize the structure contained in the structure input file given in the -cs, -rs, or -ls options and make sure it is consistent (as shown in Fig 1, right panel). On the other hand, if the structure is "broken" (as shown in Fig 1, left panel) this could generate inconsistent results.
Generate the structure from tpr file:
gmx editconf -f md.tpr -o md.pdb

The trajectory defined in -ct, -rt, or -lt doesn't contain PBC
Visualize the trajectory given in the -ct, -rt, or -lt options and make sure the PBC has been removed (as shown in Fig 2, right panel). On the other hand, if the trajectory has not been fitted (as shown in Fig 2, left panel) this could generate inconsistent results.
Steps:
-
Generate a group that contains both molecules
gmx make_ndx -n index.ndx >1 | 12 >qAssuming 1 is the receptor and 12 is the ligand. This creates a new group (number 20 in this example)
-
remove the PBC
gmx trjconv -s md.tpr -f md.xtc -o md_noPBC.xtc -pbc mol -center -n -ur compact center: 20 (created group) output: 0 -
remove the rotation and translation with respect to the reference structure (optional)
gmx trjconv -s md.tpr -f md_noPBC.xtc -o md_fit.xtc -n -fit rot+trans fit: 20 (created group) output: 0 -
Visualization
Make sure that the trajectory is consistent (as shown in Fig 2, right panel)
-
If the process is not succesful, consider using other options like
-pbc nojump(as suggested here)

Running gmx_MMPBSA¶
Tip
- Since version 1.4.0 we have fixed the
gmx_MMPBSAinconsistencies when usingMPI. - We currently recommend the use of MPI since the computation time decreases considerably.
gmx_MMPBSA as MMPBSA.py uses the MPI only to perform the calculations, the rest of the process (i.e, Generation/conversion of Amber topologies, mutation, division of the trajectories, etc) occurs in a single thread (See Figure 3 for better reference). This means that it is not necessary to install any program (AmberTools or GROMACS) with MPI, which can be used in any circumstance, and the time required to process the data prior to the calculation depends on the system and will be the same for both versions (Serial and MPI).
Note
Note that gmx_MMPBSA processes, converts, or builds topologies from GROMACS files, so it takes slightly longer than MMPBSA.py at the same stage of the process. However, this is not really significant.
Remember
Make sure that you install the OpenMPI library
sudo apt install openmpi-bin libopenmpi-dev openssh-client
A usage example is shown below:
mpirun -np 2 gmx_MMPBSA -O -i mmpbsa.in -cs com.tpr -ci index.ndx -cg 1 13 -ct com_traj.xtc
#!/bin/sh
#PBS -N nmode
#PBS -o nmode.out
#PBS -e nmode.err
#PBS -m abe
#PBS -M email@domain.edu
#PBS -q brute
#PBS -l nodes=1:surg:ppn=3
#PBS -l pmem=1450mb or > 5gb for nmode calculation
cd $PBS_O_WORKDIR
mpirun -np 3 gmx_MMPBSA -O -i mmpbsa.in -cs com.tpr -ci index.ndx -cg 1 13 -ct com_traj.xtc > progress.log
Danger
Unfortunately, when running gmx_MMPBSA with MPI, GROMACS's gmx_mpi can't be used. This is probably because of gmx_mpi conflicts with mpirun. In any case, this is not a problem since gmx works correctly and gmx_mpi only parallels mdrun, the rest of the GROMACS tools work in a single thread. See this issue to see the output.
Warning
The nmode calculations require a considerable amount of RAM. Consider that the total amount of RAM will be:
RAMtotal = RAM1_frame * NUM of Threads
If it consumes all the RAM of the system it can cause crashes, instability or system shutdown!
Note
At a certain level, running RISM in parallel may actually hurt performance, since previous solutions are used as an initial guess for the next frame, hastening convergence. Running in parallel loses this advantage. Also, due to the overhead involved in which each thread is required to load every topology file when calculating energies, parallel scaling will begin to fall off as the number of threads reaches the number of frames.
This version is installed via pip as described above. AMBERHOME variable must be set, or it will quit with an error. An example command-line call is shown below:
gmx_MMPBSA -O -i mmpbsa.in -cs com.tpr -ci index.ndx -cg 1 13 -ct com_traj.xtc
You can found test files on GitHub
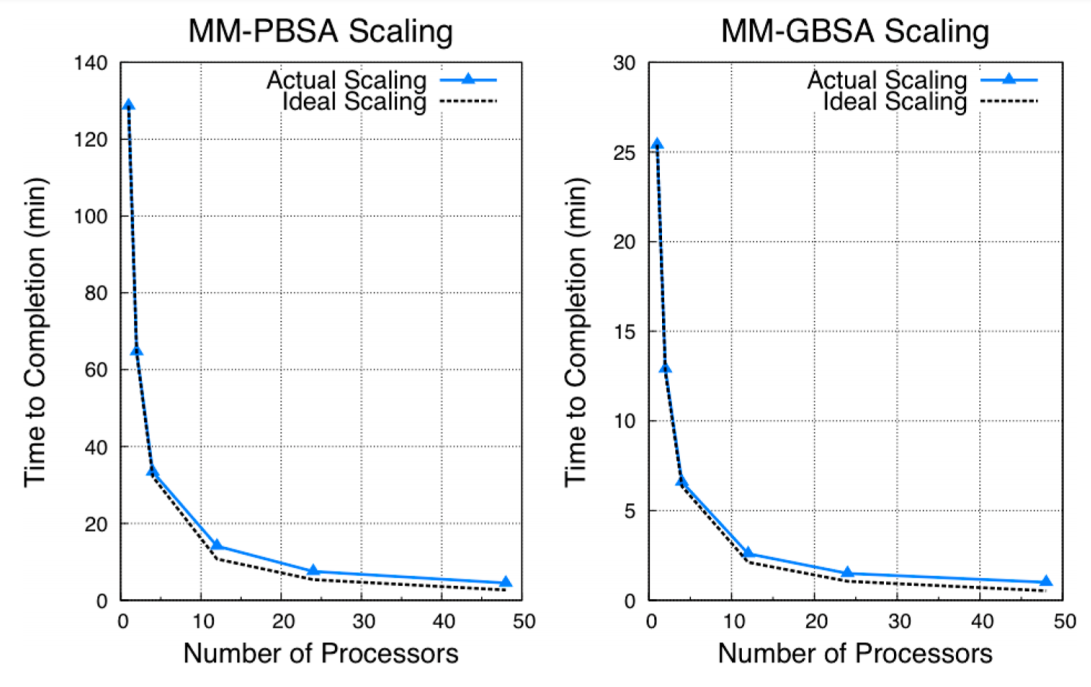
Figure 3. MPI benchmark description from MMPBSA.py paper. MMPBSA.py scaling comparison for MM-PBSA and MM-GBSA calculations on 200 frames of a 5910-atom complex. Times shown are the times required for the calculation to finish. Note that MM-GBSA calculations are ∼5 times faster than MM-PBSA calculations. All calculations were performed on NICS Keeneland (2 Intel Westmere 6-core CPUs per node, QDR infiniband interconnect)
Created: January 27, 2022 07:53:46